Что такое аплазия копчика

Синдром каудальной регрессии – редкий тяжелый врожденный порок развития дистального отдела позвоночника и спинного мозга. В иностранной литературе встречается несколько терминов, обозначающих данное патологическое состояние: сакральная или люмбосакральная агенезия, синдром каудальной дисплазии, каудальная дисгенезия. Клиническая картина (см. далее) заболевания сопровождается гипоплазией нижней половины туловища и конечностей вследствие грубого порока развития каудального отдела позвоночника и спинного мозга. В зависимости от уровня и тяжести поражения последнего наблюдается различная степень выраженности неврологического дефицита. В зависимости от уровня поражения позвоночника могут отсутствовать копчиковые, крестцовые, поясничные и даже нижнегрудные позвонки, что определяет вариант порока.
Крайне тяжелая форма каудальной регрессии называется сиреномелией, или синдромом русалки. Частота встречаемости этого летального порока – 1 на 60 тыс. новорожденных. Патогномоничным признаком данной аномалии, впервые описанной в 1961 г. Duhamel , является слияние нижних конечностей. Сращение может быть костным или в пределах мягких тканей. В большинстве слчаев сиреномелии наблюдаются агенезия почек, слепо оканчивающаяся толстая кишка, отсутствие наружных и внутренних гениталий, единственная пупочная артерия, атрезия ануса.
Этиология синдрома каудальной регрессии окончательно не выяснена. Большинство авторов причинными факторами в генезе данной патологии рассматривают сахарный диабет у матерей, генетическую предрасположенность и недостаточное кровоснабжение нижней половины тела плода – установлено, что у плодов с данной патологией кровь шунтируется через аномальный сосуд в плаценту, не осуществляя кровоснабжение каудальных структур.
Классификация синдрома каудальной регрессии. С учетом накопленного опыта и различных клинических форм первая классификация синдрома каудальной регрессии предложена в 1978 г. Renshaw, который описал четыре варианта порока:
- Тип 1 — полная или частичная односторонняя сакральная агенезия: тазовое кольцо и люмбосакральное сочленение интактны, отсутствует позвоночно-тазовая нестабильность. Односторонняя агенезия крестца приводит к перекосу таза и поясничному сколиозу, который обычно не прогрессирует и не требует хирургического лечения. У больных с типом 1 может встречаться эквиноварусная деформация стоп и неврологический дефицит в виде выпадения чувствительности в соответствии с поражением крестцовых корешков.
- Тип 2 — неполная сакральная агенезия с частичным, но двухсторонним дефектом, стабильным суставом между подвздошными костями и нормальным или гипопластичным S1 позвонком. Позвоночно-тазовое сочленение у пациентов данной группы обычно стабильно, но часто встречаются врожденные аномалии развития позвонков на фоне нарушения их формирования и слияния (полупозвонки, клиновидные и бабочковидные позвонки). Деформация позвоночника у таких пациентов приобретает прогрессирующий характер и требует хирургической коррекции. В неврологической картине отмечаются парезы или параличи нижних конечностей без нарушения чувствительности. Деформации коленных суставов и стоп выражены умеренно. Большинство больных передвигается самостоятельно.
- Тип 3 — вариабельная поясничная и полная крестцовая агенезия, при которой подвздошные кости соединяются с боковыми поверхностями последнего позвонка. У пациентов данной группы позвоночно-тазовое сочленение относительно стабильное, несмотря на отсутствие в некоторых случаях L5 позвонка. В процессе роста и развития у ребенка наблюдаются прогрессирующий кифоз или сколиоз. Неврологический статус — двигательные нарушения с уровня вертебрального дефекта. При полном отсутствии крестца ягодицы плоские, а межъягодичная складка укорочена. Отмечаются вывихи бедер, контрактуры коленных суставов и деформация стоп, требующие оперативного лечения. Пациенты не могут передвигаться без ортезов и костылей.
- Тип 4 — вариабельная поясничная и полная сакральная агенезия, когда каудальная пластинка самого нижнего позвонка располагается над сочленением подвздошных костей. Этот тип представляет собой классическую форму синдрома каудальной регрессии или люмбосакральной агенезии. Для пациентов характерно положение в позе Будды, низкий рост, выраженная диспропорциональность грудной клетки и таза. Имеется отчетливая нестабильность таза. В положении пациента сидя таз максимально приближен к грудной клетке спереди. Почти у всех пациентов развивается кифотическая или кифосколиотическая деформация. Движения в тазобедренных суставах резко ограничены из-за выраженных сгибательно-отводящих контрактур. Сгибательные контрактуры коленных суставов часто сочетаются с характерными парусовидными подколенными складками, так называемыми птеригиумами. Как правило, наблюдаются фиксированные эквиноварусные деформации стоп.
В 1996 г. Cama et al. (1996) детально изучили группу пациентов с синдромом каудальной регрессии и выделили пять категорий:
- полная агенезия крестца с нормальным или уменьшенным поперечным диаметром таза и возможным отсутствием нескольких поясничных позвонков;
- полная сакральная агенезия;
- частичная сакральная агенезия или гипоплазия крестца (сохранен S1 позвонок);
- полукрестец (hemisacrum);
- копчиковая агенезия.
В 2002 г. Guelle et al. (2002) предложили новую классификацию синдрома каудальной регрессии, связывающую клинико-рентгенологический тип порока с потенциальной возможностью самостоятельной ходьбы. На основании опыта лечения 18 пациентов выделены две группы: у пациентов первой группы (13 наблюдений) присутствовала только люмбосакральная агенезия; второй группы (5 наблюдений) — люмбосакральная агенезия сочеталась с миеломенингоцеле. В каждой из групп отмечено три типа деформации позвоночника:
- тип А — небольшой дефект меж у подвздошными костями или сращение подвздошных костей по средней линии; отсутствие одного или нескольких поясничных позвонков; каудальный позвонок сочленяется с тазом по средней линии;
- тип B — полное сращение подвздошных костей, отсутствие нескольких поясничных позвонков, каудальный позвонок сочленяется с одной из подвздошных костей;
- тип С — полное сращение подвздошных костей между собой, отсутствие всех поясничных позвонков, значительный дефект между интактным грудным позвонком и тазом.
Все пациенты первой группы с деформациями типа А и частично типа В имели хорошую перспективу к самостоятельной ходьбе после хирургической коррекции деформаций нижних конечностей. Оставшиеся больные (вторая группа; тип С и частично тип В первой группы) самостоятельно передвигаться не могли, показаниями к корригирующим вмешательствам являлись невозможность сидения и/или ношения ортопедической обуви и ортезов.
Клиника. У пациентов с синдромом каудальной регрессии отмечается низкий рост, что обусловлено укорочением туловища и конечностей. Наиболее яркими клиническими проявлениями данного синдрома являются сужение и гипоплазия таза, гипотрофия нижних конечностей, врожденные вывихи бедер, сгибательные контрактуры тазобедренных и коленных суставов, эквиноварусные деформации стоп. В большинстве описанных наблюдений данная аномалия сочетается с пороками других органов и систем, что требует привлечения к лечению пациентов специалистов различного профиля. Данный врожденный дефект может сопровождаться рядом аномалий со стороны:
- центральной нервной системы (миеломенингоцеле, гидроцефалия, мальформация Арнольда-Киари, голопрозэнцефалия),
- сердца (дефект межжелудочковой перегородки);
- желудочно-кишечного тракта – трахео-эзо-фагеальный свищ, дефект передней брюшной стенки, паховая грыжа, мальротация кишечника, атрезия двенадцатиперстной кишки, атрезия прямой кишки;
- мочеполового тракта – уретеро-гидронефроз, мочепузырный рефлюкс, экстрофия мочевого пузыря, ректовагинальный и ректоуретеральный свищи, подковообразная почка, гипоспадия, атрезия уретры, транспозиция наружных гениталий, крипторхизм).
В неврологической картине заболевания наблюдаются глубокиепарезы или плегия нижних конечностей, мышечная гипотония, угнетение сухожильных рефлексов. Выраженность неврологического дефицита прямо коррелирует с уровнем обрыва спинного мозга и корешков. Нередко агенезия какого‑либо отдела позвоночника входит в состав генетических синдромов: OEIS-комплекс (омфалоцеле, экстрофия клоаки, атрезия ануса, пороки развития крестца), VATER‑синдром (вертебральные аномалии, атрезия ануса, трахеопищеводный свищ, атрезия пищевода, аномалии почек).
Лечение. Подход к лечению пациентов с синдромом каудальной регрессии должен быть комплексным, этапным и выбор в пользу хирургического или консервативного лечения решается строго индивидуально. Ведение данной группы пациентов ставит перед врачом ряд кардинальных задач: устранение позвоночно-тазовой нестабильности, коррекцию деформаций нижних конечностей, лечение осложнений, связанных с пороками развития других органов и систем с привлечением соответствующих специалистов.Тактика ведения пациентов с позвоночно-тазовой нестабильностью различна. Многие авторы придерживаются активной хирургической тактики и считают наличие нестабильности показанием к реконструктивной операции и инструментальной фиксации с целью освобождения верхних конечностей как опоры для нестабильного туловища, защиты внутренних органов от компрессии и деформации, создания стабильного позвоночно-тазового комплекса. В целом, отмечены положительные результаты позвоночно-тазового соединения.
В оперативном лечении вывихов бедер применяется раннее открытое вправление бедра, подвертельная корригирующая остеотомия бедренной кости с остеотомией таза или без нее. Сгибательные контрактуры коленных суставов представляют особые трудности для хирургического лечения и склонны к рецидивам даже после полной коррекции. Для устранения контрактур коленных суставов используют задний релиз с Z-образной пластикой кожной подколенной складки с наложением дистракционного аппарата и постепенным разгибанием сустава в сочетании с надмыщелковой разгибательной остеотомией бедренной кости. Guille et al. считают, что корригирующие операции на нижних конечностях должны выполняться у пациентов, потенциально способных к самостоятельному передвижению. У остальных эти операции применяют с целью облегчения сидения в коляске, ношения обуви и ортезов. Для устранения деформаций стоп и придания им опороспособности выполняют трехсуставной артродез с последующим снабжением ортезами. В отношении показаний к ампутациям нижних конечностей многие авторы считают возможным выполнение двусторонней подвертельной ампутации или дезартикуляции на уровне коленных суставов при тяжелых деформациях с последующим протезированием нижних конечностей, что позволяет пациенту уверенно сидеть без опоры на верхние конечности и самостоятельно передвигаться.
Литература: 1. статья «Cиндромом каудальной регрессии» С.В. Виссарионов, И.В. Казарян (Научно-исследовательский детский ортопедический институт им. Г.И. Турнера, Санкт-Петербург), статья опубликована в журнале «ХИРУРГИЯ ПОЗВОНОЧНИКА» 2/2010 (С. 50–55) [►]. 2. статья «Лечение пациентов с синдромом каудальной регрессии» С.В. Виссарионов, И.В. Казарян, С.М. Белянчиков (Научно-исследовательский детский ортопедический институт им. Г.И. Турнера, Санкт-Петербург), статья опубликована в журнале «ХИРУРГИЯ ПОЗВОНОЧНИКА» 3/2011 (С. 56–59) [►].
Источник
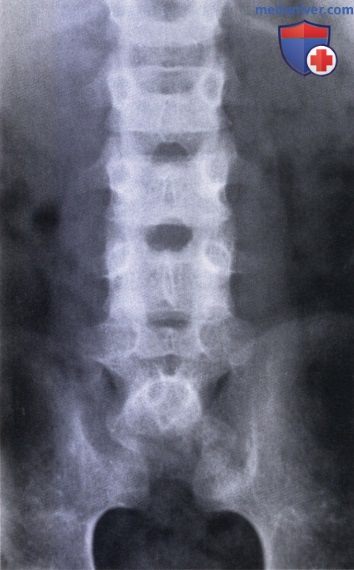
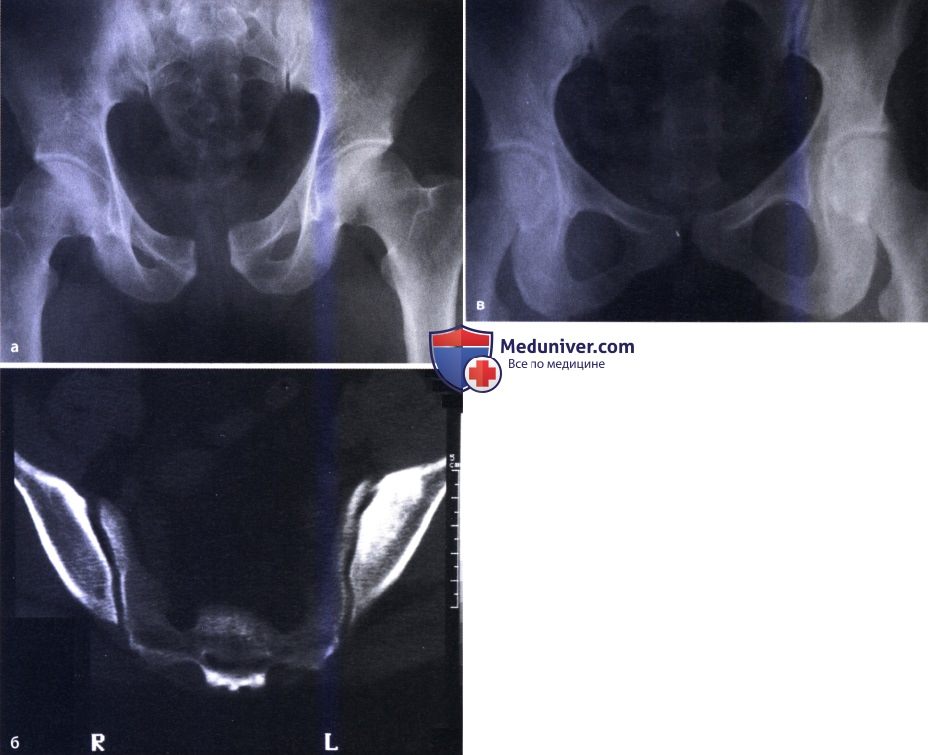
Агенезия крестца, агенезия симфиза, симфизеолиз: атлас фотографийТермином «агенезия крестца» описывается группа пороков развития, характеризующаяся отсутствием различных сегментов каудального отдела позвоночника. Агенезия крестца — это врожденная нечастая аномалия позвоночника, встречающаяся с частотой один случай на 25000 живорожденных. Частота этого порока у детей, рожденных у матерей, страдающих инсулинозависимым сахарным диабетом, составляет 1%. Внешний вид и состояние пациента зависят от распространенности поражения позвоночника и степени неврологического дефицита. У пациентов с агенезией крестца страдают моторные функции ниже уровня нормально сформированного позвоночника, также как и у пациентов с миеломенингоцеле. Ниже уровня пораженных позвонков страдают и сенсорные функции. В тяжелых случаях может полностью отсутствовать весь поясничный и даже нижнегрудной отдел позвоночника. Реншоу (Renshaw) классифицировал заболевания в зависимости от объема сохранившейся части крестца и в соответствии с характером сочленения между позвоночником и костями таза. • Тип 1 — частичная или полная односторонняя агенезия крестца; позвоночно-тазовое сочленение обычно стабильное. Нарушение чувствительности зависит от степени сохранения крестцовых корешков. Обычно развивается непрогрессирующий сколиоз. • Тип 2 — частичная агенезия крестца с двусторонним симметричным дефектом, крестцовые позвонки нормальные или с признаками гипоплазии, сочленение между S1 позвонком и подвздошными костями стабильное. • Тип 3 — различные формы агенезии поясничных позвонков и полная агенезия крестца, когда подвздошные кости сочленяются с краем самого нижнего из имеющихся позвонков. • Тип 4 — различные формы агенезии поясничных позвонков и полная агенезия крестца, при которых самый нижний позвонок находится выше сросшихся друг с другом подвздошных костей либо над их амфиартрозом.
– Также рекомендуем “Незаращение дуги позвонка (spina bifida occulta): атлас фотографий” Редактор: Искандер Милевски. Дата публикации: 23.7.2020 |
Источник
Системные аномалии развития опорно-двигательного аппарата
- Ахондрогенез – один из наиболее тяжелых видов хондродисплазий. Проявляется непропорционально большой головой, запавшей спинкой носа, выраженной микромелией с резким укорочением туловища. Выявляются отсутствие оссификации тел позвонков, костей таза, различная степень оссификации костей черепа, короткие ребра, переломы ребер, резкое укорочение и расширение длинных трубчатых костей. Оссификация лобковых и седалищных костей выражена слабо или отсутствует. Тип наследования – аутосомно-рецессивный.
- Ахондроплазия (син.: хондродисплазия, хондродистрофия врожденная, хондрогенез несовершенный) – в основе процесса лежит нарушение процесса энхондрального остеогенеза, тогда как периостальное окостенение практически не изменено. Хрящ сформирован, но резко гипоплазирован. Признаки: укорочение проксимальных отделов конечностей (ризомелическая микромелия), плюсневых и пястных костей, фаланг пальцев. Кисти широкие и имеют характерную форму, пальцы в виде трезубца, изодактилия. Постоянны микроцефалия, дисплазия лицевого черепа, (гипоплазия средней части лица, выступающие лобные бугры, седловидный нос с узкими носовыми ходами, иногда прогнатия), изменения костей таза (укорочение крыльев подвздошных костей, сужение крестцово-подвздошного сочленения, уплощение крыши и неправильные контуры вертлужной впадины). Характерны изменения позвоночника. При относительно нормальной его длине наблюдается симптом сужения расстояния между корнями дужек поясничных позвонков, нарастающий в каудальном направлении. Часто отмечается поясничный кифоз, а позднее может развиться и лордоз. Тип наследования – аутосомно-доминантный.
- Гисплазия фиброзная (син.: болезнь Брайцева – Лихтенштейна, болезнь Джеффи – Лихтенштейна) – замещение компактного слоя кости аваскулярной фиброзной тканью, обусловленное нарушением эмбриогенеза кости на соединительнотканной стадии. При рентгенологическом исследовании определяются четко отграниченные очаги просветления кости различной величины и формы, соответствующие участкам недифференцированной волокнистой ткани. По локализации различают монооссальную, полиоссальную и регионарную формы, а по характеру изменений в кости – очаговую и диффузную формы. Преимущественно поражаются трубчатые кости конечностей, кости черепа. Наблюдаются патологические переломы.
- Остеогенез несовершенный (син.: костеобразование несовершенное) – в основе лежит дефект костеобразования, связанный с недостаточностью мезенхимы. Характеризуется патологической ломкостью костей в сочетании с другими аномалиями. Поражаются преимущественно длинные трубчатые кости. Микроскопически отмечается истончение кортикального слоя трубчатых костей, разрежение и истончение костных балок губчатого вещества. В ряде случаев выявлена дисплазия эпифизов бедренных, большеберцовых и плечевых костей.
- Остеопетроз (синдром Albers – Schonberg, болезнь мраморная, остеосклероз врожденный, дисплазия гиперостотическая) – генерализованный диффузный остеосклероз, характеризуется неравномерным, широко распространенным гиперостозом, захватывающим одновременно большое количество костей с переходом процесса на костномозговой канал. Обусловлен нарушением соотношения между новообразованной и рассасывающейся костной тканью. Характерны увеличенная плотность длинных трубчатых костей, сужение костномозговой полости, замещение костного мозга костно-хрящевой тканью. Утолщенные кости имеют мраморно-белый вид, хрупкие, ломкие; часты патологические пере- ломы. В процесс вовлекаются и кости черепа. Аутосомно-рецессивное наследование.
- Остеопойкилоз (син.: остеопойкилия гиперпластическая, «кости пятнистые», остеопатия склерозирующая рассеянная врож- денная) – мелкоочаговые эндостальные скопления склерозированной костной ткани. Чаще поражаются кости кисти, стопы, запястья, предплюсны и метафизарные отделы длинных трубчатых костей.
- Пикнодизостоз – генерализованный склероз костей без нарушения их формы и сужения костномозговой полости. Характерны также гипоплазия дистальных фаланг, тупой угол нижней челюсти, широкие венечные швы.
- Хондродисплазия точечная (син.: хондродисплазия пятнистая, хондродистрофия точечная, эпифизы испещренные, хондродистрофия кальцифицирующая врожденная, дисплазия эпифизар- ная пятнистая) – появление множественных точечных (пятнистых) кальцификатов в эпифизах, периартикулярных тканях и зоне роста костей. Может наблюдаться тотальное поражение всех костей (генерализованная форма) или отдельных костей. Характерные признаки: диспластическое лицо, гипоплазия хрящей носа, укорочение конечностей, сгибательные контрактуры суставов, деформации стоп, позвоночного столба, изменения кожи (эритродермия, ихтиоз), катаракты. Различают несколько форм: летальную ризомелическую, форму Конради – Хюнерманна, Х-сцепленную доминантную и др.
- Экзостозы хрящевые множественные (син.: хондродисплазия экзостозная, экзостозы хрящевые юношеские, экзостозы костно-хрящевые множественные, аклазия диафизарная, хондродисплазия деформирующая, хондроматоз кости наружный, болезнь Эренфрида) – порок развития пластинки роста, выражающийся в появлении хрящевых и костно-хрящевых разрастаний (экзостозов) в эпиметафизарных отделах костей. Чаще поражаются кости плеча, предплечья, бедра и голени. Приводит к различного рода деформациям конечностей. Иногда сочетается с другими аномалиями – расщелиной неба, синдактилией, ангиоматозом. Аномалии развития опорно-двигательного аппарата
- Энхондроматоз (син.: синдром Ollier, hemichondrodysplasia) – наличие в метафизах и диафизах длинных трубчатых костей или теле плоских костей очагов эмбриональной хрящевой ткани, соответственно которым при рентгенографическом исследовании выявляются различной формы и размеров очаги просветления. В основе лежит замедление и извращение оссификации эмбрионального хряща, заключающееся в отсутствии замещения хрящевого скелета костной тканью. Различают по распространенности процесса монооссальную, олигооссальную и полиоссальную формы, а по локализации – 4 формы: акроформу (поражение кистей и стоп), мономелическую (поражение костей одной конечности с прилежащей частью тазового или плечевого пояса), одностороннюю или преимущественно одностороннюю и двустороннюю формы. Проявляется укорочением трубчатых костей, уплощением их с деформацией и искривлением, наблюдаются переломы, иногда экзостозы. Рентгенологически – укорочение длинных и коротких трубчатых костей, множественные односторонние энхондромы. Предполагается аутосомно-доминантное наследование.
Источник